Samtools (sometimes) can't find reference
Hi everyone,
I am using samtools through the Swiss Army Knife, and even though I have run virtually the same code in the past, I am now getting the following error:
[W::find_file_url] Failed to open reference "https://www.ebi.ac.uk/ena/cram/md5/974dc7aec0b755b19f031418fdedf293": Input/output error
[E::fai_build3_core] Failed to open the file /sbgenomics/workspaces/43f03b05-9336-4ffa-870c-e9ebdcd9d943/tasks/129fd9d0-a2c4-4a70-9933-8ec845fd5ea5/SAMtools_View___1_9/GRCh38_primary_assembly_plus_ebv_alt_decoy_hla.fasta
[E::refs_load_fai] Failed to open reference file '/sbgenomics/workspaces/43f03b05-9336-4ffa-870c-e9ebdcd9d943/tasks/129fd9d0-a2c4-4a70-9933-8ec845fd5ea5/SAMtools_View___1_9/GRCh38_primary_assembly_plus_ebv_alt_decoy_hla.fasta'
[E::cram_get_ref] Failed to populate reference for id 20
[E::cram_decode_slice] Unable to fetch reference #20:8212965-8214026
[E::cram_next_slice] Slice decode failure
[main_samview] retrieval of region 0 failed due to truncated file or corrupt BAM index file
[W::find_file_url] Failed to open reference "https://www.ebi.ac.uk/ena/cram/md5/974dc7aec0b755b19f031418fdedf293": Input/output error
[E::fai_build3_core] Failed to open the file /sbgenomics/workspaces/43f03b05-9336-4ffa-870c-e9ebdcd9d943/tasks/129fd9d0-a2c4-4a70-9933-8ec845fd5ea5/SAMtools_View___1_9/GRCh38_primary_assembly_plus_ebv_alt_decoy_hla.fasta
[E::refs_load_fai] Failed to open reference file '/sbgenomics/workspaces/43f03b05-9336-4ffa-870c-e9ebdcd9d943/tasks/129fd9d0-a2c4-4a70-9933-8ec845fd5ea5/SAMtools_View___1_9/GRCh38_primary_assembly_plus_ebv_alt_decoy_hla.fasta'
[E::cram_get_ref] Failed to populate reference for id 20
[E::cram_decode_slice] Unable to fetch reference #20:8213876-8214909
Something similar happened a few days back, but it eventually stopped throwing the same error after I literally copy pasted the same code I was running from somewhere else and retried, which made me think I had some hidden characters in the script that were messing up the execution. I have double-checked and I have none.
I relaunched the same script that worked well last week, and it completed successfully since it literally copied the output from the previous success (I assume SAK is aware that the script hadn't changed?). However, if I download that same script and change its name before reuploading it, it fails with the error above when I launch it.
As an aside, I am using SAK 4.5.0 explicitly due to the error that was discovered here (https://community.dnanexus.com/s/question/0D5t0000045Gx4GCAS/extract-multiple-regions-from-cram-with-samtools-view-on-the-rap).
Can anyone help me figure out how to solve this? I am happy to share code if necessary.
Cheers,
Fran
Comments
7 comments
This is a hard one without seeing the actual code. You may need to follow up by sending the same question above to ukbiobank-support@dnanexus.com. If you share the project with org-support, they could see the log file.
For what I know, 1) there are some corrupted CRAM in WGS. The UKB and our data teams are working on getting the right files. It could be those files. If you can send me the file id, I could verify that. 2), as you point out, the current version of qctools has out of memory problem, so we recommended people using older version of SAK which has older version of qctools if users want to use qctools. I could tell our engineer to update latest SAK to use older qctools, but we aren't sure if there are other bugs in old version of tools, so we prefer to update to newer (future) version of qctools once the out of memory bug is fixed.
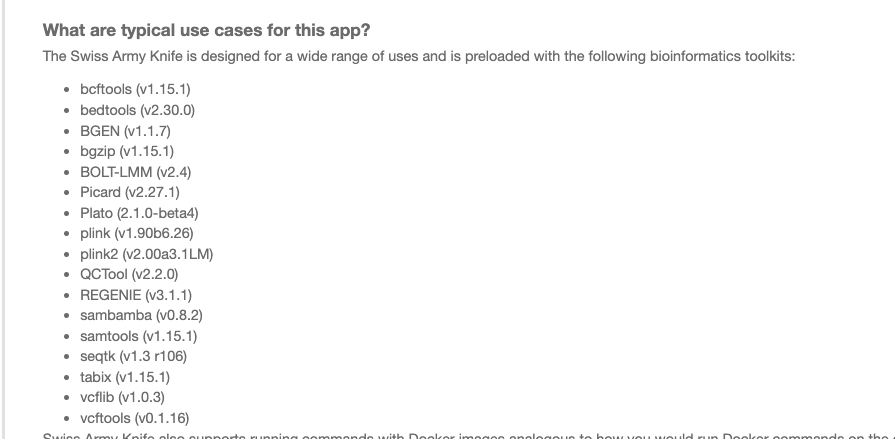
There are several version of SAK. We update it reguarly once the new version of underneath tools are available (e.g. new version of samtools, etc). Each version would have a fixed set of tools underneath. You can click on link behind executable to check the version of SAK. You can also see the README of such app to see versions of all tools underneath.
?
Thanks, @Chai Fungtammasan? . I doubt it is an issue with the CRAMs themselves, since I have successfully run tons of scripts on all of them already (including, as I said, exactly this same one). It feels to me that whatever corruption arises, if any, only happens when certain instances are assigned to the job. Incidentally, I always use dxfuse to stream the files, in case that helps narrow down the possible cause
Quick update. Just to validate my instance-specific theory on the issue, I ran the same script (on a single sample) only changing the type of instance requested (from mem1_ssd1_v2_x16 to mem1_ssd1_v2_x36) and it worked perfectly fine
Completed the entire job without issue for all samples when requesting mem1_ssd1_v2_x36.
In the "info" option of log file, did you see out of memory or full storage capacity for the job that you use mem1_ssd1_v2_x16?
No, the errors occurred from the very start of the execution. I am now running exactly the same script again on mem1_ssd1_v2_x16 (I terminated the mem1_ssd1_v2_x36 executions since they had been assigned to on-demand nodes and were becoming too expensive) and it's working without issue (which, again, makes me think it is an instance-specific problem)
One of the running ones is job-GJQVkG0JbvpyxK5v3xgXX1kZ in case you want to check it. The log of a problematic one would look like job-GJPzkBQJbvpjj6Y3PBYVzg5x. I can send its log file if you think it would be useful
I can't think of any reason why some instance would have problem while the other don't. Could you share your project with org-support and send E-mail to ukbiobank-support@dnanexus.com, so they could investigate?
Please sign in to leave a comment.