I would like to somatic mutation analysis with UKB Exome sequence data. My aim find to CHIP carriers in my cohort. I am planning to use Mutectcaller (Parabricks accelerated) app to call somatic mutations on UKB Exome sequence data. But I don't have BAM file which input file. I would like to convert CRAM to BAM using Swiss Army Knife. When I clicked run in the Swiss Army Knife, I couldn't select input file as CRAM. How can I run this tool using CRAM file as input file?
To convert cram to bam, you will need cram and reference file. I did an example testing run via SAK GUI on the "/Bulk/Exome sequences/Exome OQFE CRAM files/":
A) I clicked on Input files and selected two files --> CRAM file, and reference fa genome from the helper files folder (just click on the arrow to get the nested folders).
B) I then specified example cram2bam samtools command to be executed "samtools view -T GRCh38_full_analysis_set_plus_decoy_hla.fa -b 1xyztyf_23143_0_0.cram > output.bam" and the job finished successfully.
0
Permanently deleted user
Thank you for your response. I started analysis but I received an error. :(
Overview
-----------
Reporter: Burcu Çevik
Executable: app-GKyyzJQ951j4Bkfq4jFkGX1K
Failure Message: Error while running the command (please refer to the job log for more information).
I suspect this might have some copy and paste issue, where the "samtools command" is missing the "s" character. Make sure you run "samtools ...", not "amtools ...".
0
Permanently deleted user
Thank you for your suggestions. I have tried to rerun. Following your recommendations, I successfully was able to get result.
I would like to run a batch which includes many jobs to convert .cram files to .bam files. I tried to do that using batch mode but I could not configure batch run. I'm sending a screenshot of the problem in the attachment. Tool doesn't allow user to select two or more input files within the same job. If I select two input files (.cram and .fa) for one job, second input file is located within other row. There is not another column for GRCh38_full_analysis_set_plus_decoy_hla.fa file. I can not start analysis in this way.
Could you please help me to run multiple CRAM files at the same time?
Comments
8 comments
To convert cram to bam, you will need cram and reference file. I did an example testing run via SAK GUI on the "/Bulk/Exome sequences/Exome OQFE CRAM files/":
A) I clicked on Input files and selected two files --> CRAM file, and reference fa genome from the helper files folder (just click on the arrow to get the nested folders).
Thank you for your response. I started analysis but I received an error. :(
Overview
-----------
Reporter: Burcu Çevik
Executable: app-GKyyzJQ951j4Bkfq4jFkGX1K
Failure Message: Error while running the command (please refer to the job log for more information).
Launched By: user-cevikb
Launched Timestamp: 03/02/2023 1:38 pm
Total Run Time: 2m
Job ID: job-GQ07k48JbBpZByvQ22z7Q925
Job Link: /projects/GKQbbYQJbBpk5YXf0JpzGXKK/monitor/job/GQ07k48JbBpZByvQ22z7Q925
Failure from origin-job
--------------------------------
{
"id": "job-GQ07k48JbBpZByvQ22z7Q925",
"name": "Swiss Army Knife",
"function": "main",
"stage": null,
"analysis": null,
"executable": "app-GKyyzJQ951j4Bkfq4jFkGX1K",
"executableName": "swiss-army-knife",
"failureReason": "AppError",
"failureMessage": "Error while running the command (please refer to the job log for more information)."
}
Origin-job Inputs
--------------------
undefined
View log of failed sub-job
--------------------------------
Logging initialized (priority)
Downloading bundled file resources.tar.gz
>>> Unpacking resources.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file qctool.tar.gz
>>> Unpacking qctool.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file plato.tar.gz
>>> Unpacking plato.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file bedtools.tar.gz
>>> Unpacking bedtools.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file htslib.tar.gz
>>> Unpacking htslib.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file java.tar.gz
>>> Unpacking java.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file plink.tar.gz
>>> Unpacking plink.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file r.tar.gz
>>> Unpacking r.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file sambamba.tar.gz
>>> Unpacking sambamba.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file seqtk.tar.gz
>>> Unpacking seqtk.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file vcflib.tar.gz
>>> Unpacking vcflib.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file vcftools.tar.gz
>>> Unpacking vcftools.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file plink2.tar.gz
>>> Unpacking plink2.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file regenie.tar.gz
>>> Unpacking regenie.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file bolt-lmm_asset.tar.gz
>>> Unpacking bolt-lmm_asset.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file bgen.tar.gz
>>> Unpacking bgen.tar.gz to /
tar: Removing leading `/' from member names
dxpy/0.340.1 (Linux-5.4.0-1096-aws-x86_64-with-glibc2.29)
bash running (job ID job-GQ07k48JbBpZByvQ22z7Q925)
downloading file: file-G572Pj8JykJZ52BP4z6GqB21 to filesystem: /home/dnanexus/in/in/0/GRCh38_full_analysis_set_plus_decoy_hla.fa
downloading file: file-G55jg28JykJfF2KJ4yPQFz40 to filesystem: /home/dnanexus/in/in/1/1000229_23143_0_0.cram.crai
Using dxfuse version v1.0.0
The log file is located at /root/.dxfuse/dxfuse.log
starting fs daemon
wait for ready
Daemon started successfully
Downloading files using 4 threads+ [[ '' == '' ]]
+ eval 'samtools view -T GRCh38_full_analysis_set_plus_decoy_hla.fa -b 1000229_23143_0_0.cram > output.bam'
++ samtools view -T GRCh38_full_analysis_set_plus_decoy_hla.fa -b 1000229_23143_0_0.cram
[E::hts_open_format] Failed to open file "1000229_23143_0_0.cram" : No such file or directory
samtools view: failed to open "1000229_23143_0_0.cram" for reading: No such file or directory
END_LOG
Hi @Burcu Çevik?, one line in the job log you shared says
"downloading file: file-G55jg28JykJfF2KJ4yPQFz40 to filesystem: /home/dnanexus/in/in/1/1000229_23143_0_0.cram.crai".
It appears you provided crai file (index) of the given cram file, instead of the cram file itself. You may try to rerun with 1000229_23143_0_0.cram.
Hi {@005t0000006BZL2AAO}? ,
I have tried rerun with .cram but I received an error.
Overview
-----------
Reporter: Burcu Çevik
Executable: app-GKyyzJQ951j4Bkfq4jFkGX1K
Failure Message: Error while running the command (please refer to the job log for more information).
Launched By: user-cevikb
Launched Timestamp: 03/02/2023 3:27 pm
Total Run Time: 2m
Job ID: job-GQ09GG8JbBpb85pyp9QZ42f3
Job Link: /projects/GKQbbYQJbBpk5YXf0JpzGXKK/monitor/job/GQ09GG8JbBpb85pyp9QZ42f3
Failure from origin-job
--------------------------------
{
"id": "job-GQ09GG8JbBpb85pyp9QZ42f3",
"name": "Swiss Army Knife",
"function": "main",
"stage": null,
"analysis": null,
"executable": "app-GKyyzJQ951j4Bkfq4jFkGX1K",
"executableName": "swiss-army-knife",
"failureReason": "AppError",
"failureMessage": "Error while running the command (please refer to the job log for more information)."
}
Origin-job Inputs
--------------------
undefined
View log of failed sub-job
--------------------------------
Logging initialized (priority)
Downloading bundled file resources.tar.gz
>>> Unpacking resources.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file qctool.tar.gz
>>> Unpacking qctool.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file plato.tar.gz
>>> Unpacking plato.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file bedtools.tar.gz
>>> Unpacking bedtools.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file htslib.tar.gz
>>> Unpacking htslib.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file java.tar.gz
>>> Unpacking java.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file plink.tar.gz
>>> Unpacking plink.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file r.tar.gz
>>> Unpacking r.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file sambamba.tar.gz
>>> Unpacking sambamba.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file seqtk.tar.gz
>>> Unpacking seqtk.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file vcflib.tar.gz
>>> Unpacking vcflib.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file vcftools.tar.gz
>>> Unpacking vcftools.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file plink2.tar.gz
>>> Unpacking plink2.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file regenie.tar.gz
>>> Unpacking regenie.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file bolt-lmm_asset.tar.gz
>>> Unpacking bolt-lmm_asset.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file bgen.tar.gz
>>> Unpacking bgen.tar.gz to /
tar: Removing leading `/' from member names
dxpy/0.340.1 (Linux-5.4.0-1096-aws-x86_64-with-glibc2.29)
bash running (job ID job-GQ09GG8JbBpb85pyp9QZ42f3)
downloading file: file-G5624g8JykJqJ3p94qzbQv76 to filesystem: /home/dnanexus/in/in/0/6000001_23143_0_0.cram
downloading file: file-G572Pj8JykJZ52BP4z6GqB21 to filesystem: /home/dnanexus/in/in/1/GRCh38_full_analysis_set_plus_decoy_hla.fa
Using dxfuse version v1.0.0
The log file is located at /root/.dxfuse/dxfuse.log
starting fs daemon
wait for ready
Daemon started successfully
Downloading files using 4 threads+ [[ '' == '' ]]
+ eval 'amtools view -T GRCh38_full_analysis_set_plus_decoy_hla.fa -b 6000001_23143_0_0.cram > output.bam'
++ amtools view -T GRCh38_full_analysis_set_plus_decoy_hla.fa -b 6000001_23143_0_0.cram
/home/dnanexus/job-GQ09GG8JbBpb85pyp9QZ42f3.code.sh: line 69: amtools: command not found
END_LOG
From the lines in your error log,
"Downloading files using 4 threads+ [[ '' == '' ]]
+ eval 'amtools view -T GRCh38_full_analysis_set_plus_decoy_hla.fa -b 6000001_23143_0_0.cram > output.bam'
++ amtools view -T GRCh38_full_analysis_set_plus_decoy_hla.fa -b 6000001_23143_0_0.cram
/home/dnanexus/job-GQ09GG8JbBpb85pyp9QZ42f3.code.sh: line 69: amtools: command not found",
I suspect this might have some copy and paste issue, where the "samtools command" is missing the "s" character. Make sure you run "samtools ...", not "amtools ...".
Thank you for your suggestions. I have tried to rerun. Following your recommendations, I successfully was able to get result.
Awesome!
Hi @Ondrej Klempir? ,
I would like to run a batch which includes many jobs to convert .cram files to .bam files. I tried to do that using batch mode but I could not configure batch run. I'm sending a screenshot of the problem in the attachment. Tool doesn't allow user to select two or more input files within the same job. If I select two input files (.cram and .fa) for one job, second input file is located within other row. There is not another column for GRCh38_full_analysis_set_plus_decoy_hla.fa file. I can not start analysis in this way.
Could you please help me to run multiple CRAM files at the same time?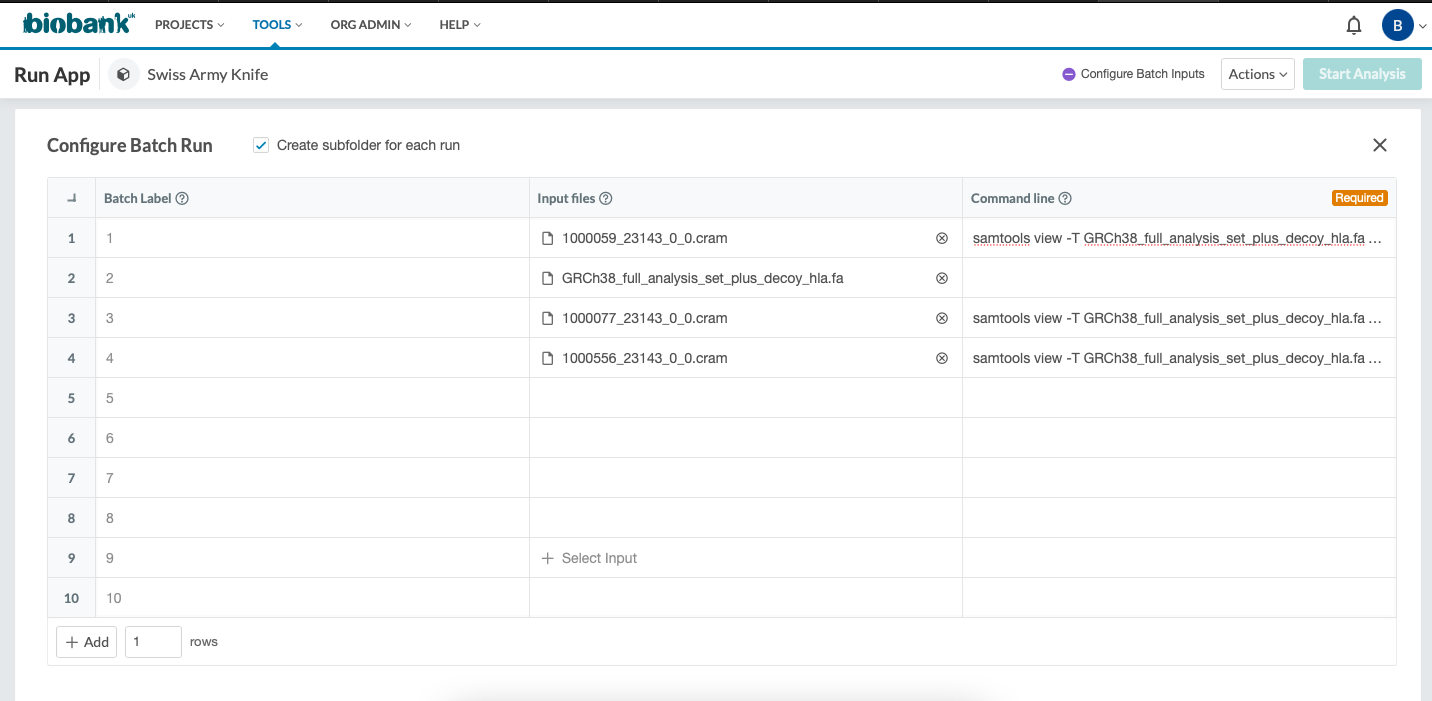
Please sign in to leave a comment.