How to fix QCtool code to filter bgen file?
I am trying to use QCtool within SwissArmyKnife on the UKB RAP to filter bgen files down to specific SNPs. I am practicing with a single bgen. The below ran for 15 minutes, but then threw an error. How can I fix the code?
Downloading files using 4 threads+ [[ '' == '' ]]
+ eval 'qctool -g ukb22828_c1_b0_v3.bgen -og subsetted.bgen -incl-rsids -incl-variants-matching rsid~rs54%'
++ qctool -g ukb22828_c1_b0_v3.bgen -og subsetted.bgen -incl-rsids -incl-variants-matching rsid~rs54%
Welcome to qctool (version: 2.2.0, revision: unknown) (C) 2009-2020 University of Oxford Opening genotype files : [ ] (0/1,0.0s,0.0/s) Opening genotype files : [******************************] (1/1,0.2s,4.1/s) Opening genotype files : [******************************] (1/1,0.2s,4.1/s) ======================================================================== Input SAMPLE file(s): Output SAMPLE file: "(n/a)". Sample exclusion output file: "(n/a)". Input GEN file(s): (not computed) "snp-id-data-filtered:ukb22828_c1_b0_v3.bgen (bgen v1.2; 487409 unnamed samples; zlib compression)" (total 1 sources, number of snps not computed). Number of samples: 487409 Output GEN file(s): "subsetted.bgen" Output SNP position file(s): (n/a) Sample filter: . SNP filter: rsid~rs54%. # of samples in input files: 487409. # of samples after filtering: 487409 (0 filtered out). ======================================================================== terminate called after throwing an instance of 'boost::exception_detail::clone_impl<boost::exception_detail::error_info_injector<boost::bad_get> >' what(): boost::bad_get: failed value get using boost::get /home/dnanexus/job-GGG0q40JkvjQVKqV8949V0B5.code.sh: line 69: 16714 Aborted qctool -g ukb22828_c1_b0_v3.bgen -og subsetted.bgen -incl-rsids -incl-variants-matching rsid~rs54%
Comments
8 comments
I was able to reproduce this issue with the recent version of qctool. It worked well with an example small bgen, however, it failed when inputting a larger UKB bgen file. This seems to me as a bug in the qctool. We informed DNAnexus dev team about this.
As a workaround, you might try running an older version of Swiss Army Knife. I tested 4.1.1 and it seems to me that this is working without errors for UKB bgen.
Oh excellent, thanks so much for that Ondrej! I will try running that on 4.1.1 now.
I was also wondering if there is a way to run this command over multiple bgen files at once? I.e. have all 22 chromosome bgens as the input and 22 filtered bgens as the output, while supplying just one list/external file of rsids?
Hi Ondrej - 4.1.1 is still giving me problems. I seem to be having a space issue. Is there any workaround for this?
I first tried running:
qctool -g ukb22828_c#_b0_v3 -og subsetted.bgen -incl-positions GWASsnps.txt
inputting all 22 chromosome bgens, but that threw a 'no space left on device' error.
Then I just limited the operation to one bgen file:
qctool -g ukb22828_c1_b0_v3 -og c1subsetted.bgen -incl-positions GWASsnps.txt
but I ran into the same error:
>>> Unpacking plink2.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file regenie.tar.gz
>>> Unpacking regenie.tar.gz to /
tar: Removing leading `/' from member names
Downloading bundled file bolt-lmm_asset.tar.gz
>>> Unpacking bolt-lmm_asset.tar.gz to /
tar: Removing leading `/' from member names
dxpy/0.327.1 (Linux-5.4.0-1083-aws-x86_64-with-glibc2.29)
/usr/sbin/sshd already running.
/usr/sbin/rsyslogd already running.
bash running (job ID job-GGPBK6QJkvjpqXG41FZ39ZXq)
Using dxfuse version v0.23.3
The log file is located at /root/.dxfuse/dxfuse.log
starting fs daemon
wait for ready
Daemon started successfully
downloading file: file-GGP8px0JkvjQJPY2F300gQGj to filesystem: /home/dnanexus/in/in/0/GWASsnps.txt
downloading file: file-FxY5660JkF6BB3Jq9680pjqX to filesystem: /home/dnanexus/in/in/1/ukb22828_c1_b0_v3.bgen
CPU: 11% (4 cores) * Memory: 1657/7661MB * Storage: 44/96GB * Net: 75?/1?MBps
Sep 12, 2022 1:37 PM
Downloading files using 4 threads'file-FxY5660JkF6BB3Jq9680pjqX' -> in/1/ukb22828_c1_b0_v3.bgen generated an exceptionTraceback (most recent call last):
Sep 12, 2022 1:48 PM
File "/usr/local/bin/dx-download-all-inputs", line 77, in <module>
dxpy.download_all_inputs(exclude=args.exclude, parallel=args.parallel)
File "/usr/local/lib/python3.8/dist-packages/dxpy/bindings/download_all_inputs.py", line 200, in download_all_inputs
_parallel_file_download(to_download, idir, max_num_parallel_downloads)
File "/usr/local/lib/python3.8/dist-packages/dxpy/bindings/download_all_inputs.py", line 69, in _parallel_file_download
future.result()
File "/usr/lib/python3.8/concurrent/futures/_base.py", line 437, in result
return self.__get_result()
File "/usr/lib/python3.8/concurrent/futures/_base.py", line 389, in __get_result
raise self._exception
File "/usr/lib/python3.8/concurrent/futures/thread.py", line 57, in run
result = self.fn(*self.args, **self.kwargs)
File "/usr/local/lib/python3.8/dist-packages/dxpy/bindings/download_all_inputs.py", line 49, in _download_one_file
dxpy.download_dxfile(src_file, trg_file)
File "/usr/local/lib/python3.8/dist-packages/dxpy/bindings/dxfile_functions.py", line 131, in download_dxfile
success = _download_dxfile(dxid,
File "/usr/local/lib/python3.8/dist-packages/dxpy/bindings/dxfile_functions.py", line 382, in _download_dxfile
fh.write(chunk_data)
OSError: [Errno 28] No space left on device
Low scratch storage space
I would write a submission script which would either 1) run the Swiss Army Knife separately for each chrom or 2) process bgens sequentially one by one. You should be able to provide a shell script to SAK. For 2), I would not download all the files into the worker (bgen is a large file, tens of GBs), and more access those separately.
Yeah, you will need to select an instance type with sufficient capacity of storage and memory. Here is a list of instance types:
https://dnanexus-prod-asg-dnanexusprodassets4d7ed69b-i607e894f3ya.s3.us-east-1.amazonaws.com/images/files/UKB_Rate_Card-Current.pdf
UKB bgen is a large file, tens or hundred of GBs.
Thanks so much for these answers Ondrej. So the submission script can only be run via a CLI, and not the analysis GUI on the RAP?
And thank you for explaining the instance issue to me! :)
Hi @Rachel Visontay?,
I do not have much practical experience with running SAK in GUI batch mode, but I was able to locate the following setting for SAK:
Oh thanks so much for this Ondrej! I tried specifying the batches and then running: bgenix -g ukb22828_c*_b0_v3.bgen -incl-rsids GWASsnpsRSIDS.txt > c*filtered.bgen
The error I'm encountering now is that only a single file can be specified per batch - in the below I have five files, each as inputs in a separate batch job.
But I only want 2 jobs to run (one for C21, and one for C22), but each batch requires three files as input (the rsid text file, the bgen file, and the relevant bgi file). So the error I get for the two jobs of interest is that the bgi file can't be opened...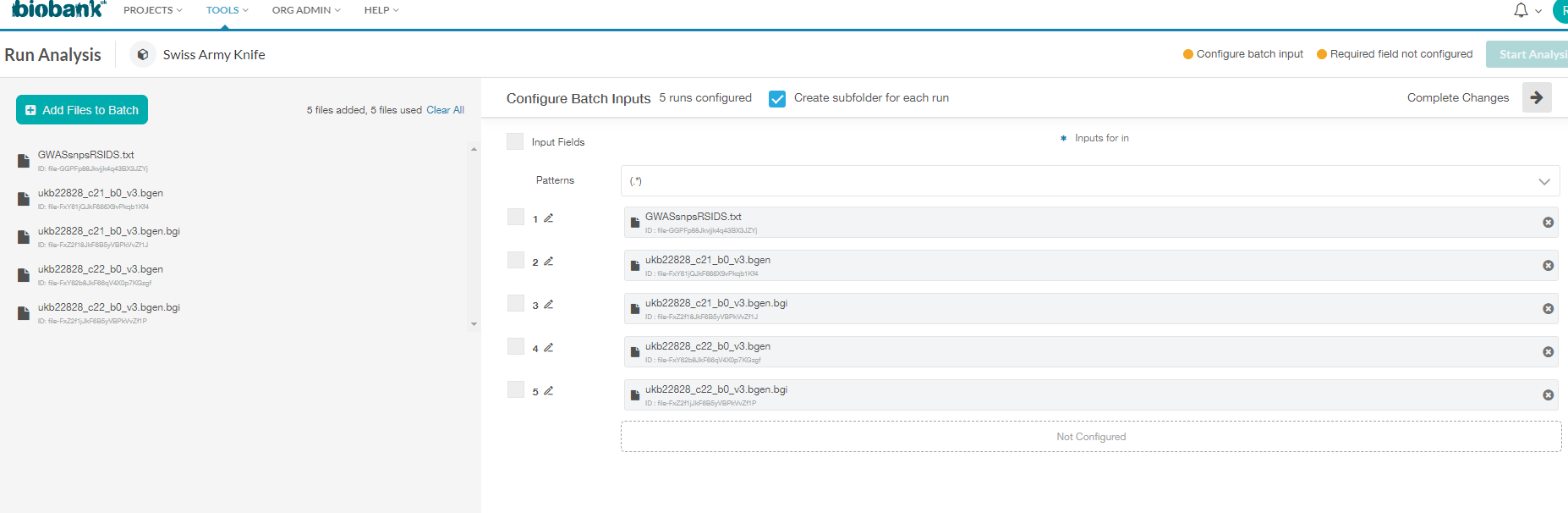
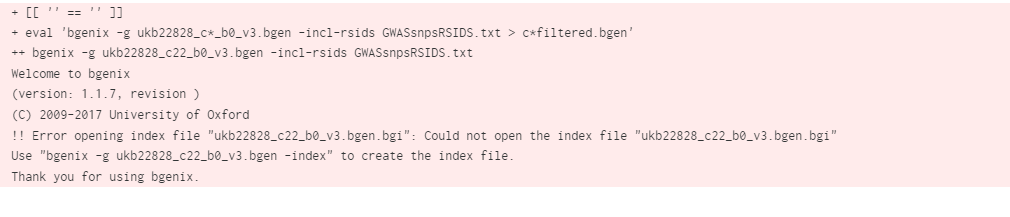 The batch example here: https://documentation.dnanexus.com/science/scientific-guides/saige-gwas-walkthrough
The batch example here: https://documentation.dnanexus.com/science/scientific-guides/saige-gwas-walkthrough
uses a different app, and allows for specifying multiple input files per batch. How would I do that for SAK?
Please sign in to leave a comment.